
Tan importante como comprender el funcionamiento de las vacunas es conocer los exhaustivos pasos y fases que han de completarse antes de su distribución al público. El siguiente apartado de la serie Vacunación relata los diferentes procedimientos, a la vez que analiza con detalle el proceso de fabricación de la vacuna del COVID19 y su posterior aprobación.
La preparación de las vacunas es otro de los temas que genera mucha controversia y que hace crecer la comunidad de antivacunas. Esto se debe a la poca confianza que a muchos les inspira el sistema de control y aprobación de las vacunas, que normalmente está en manos de organismos gubernamentales. Sin embargo, no debemos olvidar que todas las vacunas tienen que pasar obligatoriamente estrictas fases y que pasan por un control científico exhaustivo con resultados publicados en revistas científicas de prestigio.
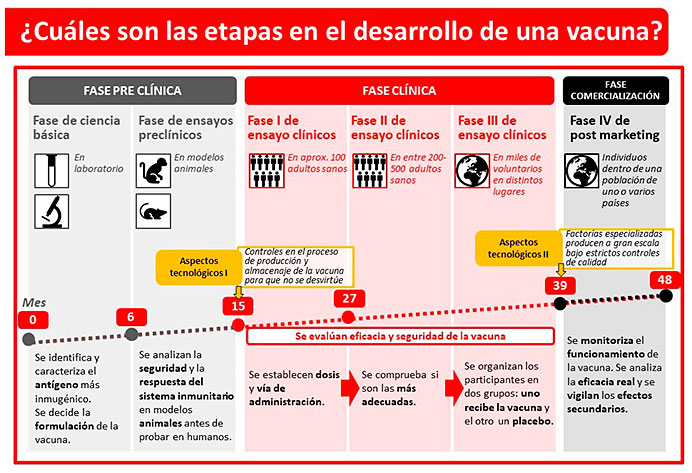
La preparación de las vacunas se puede dividir en varias etapas que pueden durar desde meses a años:

• Investigación y desarrollo. Tras un descubrimiento en particular o haber tenido una idea sobre cómo desarrollar una vacuna contra una enfermedad, en esta primera etapa estas ideas de los científicos se analizan y se estudian pormenorizadamente. El desarrollo de una vacuna suele requerir varios años de investigación de laboratorio, generalmente en una empresa del sector privado, pero a menudo implica la colaboración con investigadores de instituciones públicas.
• Fase preclínica. Antes de que una vacuna pueda probarse en personas, la empresa o institución que realiza la investigación en el laboratorio lleva a cabo pruebas en animales pequeños, como ratones, para obtener información sobre su funcionamiento y si es probable que sea segura y eficaz en humanos. En esta fase se estudia la capacidad de la vacuna para provocar una respuesta inmunitaria en estos animales pequeños, información que sirve a los investigadores para optimizar la vacuna con el fin de aumentar su eficacia.
• Solicitud de autorización de ensayo clínico. Si los resultados de la fase preclínica han sido satisfactorios, la institución que investiga la vacuna solicita el permiso para comenzar a probarla en humanos. Esto se denomina “Solicitud de autorización de ensayo clínico (SAEC)” en España (“Investigational New Drug (IND) application” en EE. UU.) y se presenta a la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) y a la Agencia Europea de Medicamentos (EMA) (la EMA es más o menos la equivalente europea a la famosa “Food and Drug Administration (FDA)” en EE.UU.). En este documento se debe explicar cómo se elabora la vacuna, cómo se prueba en animales, resumir los hallazgos de laboratorio y describir el estudio propuesto en humanos. Las instituciones competentes deben decidir entonces si se aprueba el estudio y un comité de ética de la institución donde se realizarán los ensayos en humanos debe también dar el visto bueno. Una vez aprobada, la vacuna puede probarse en tres fases en humanos.
• Ensayos clínicos: Los ensayos clínicos se llevan a cabo según los protocolos establecidos por las agencias correspondientes (EMA en Europa y FDA en EEUU). Éstas cuentan con una amplia experiencia en el diseño de ensayos clínicos, por lo que están pensados para cumplir los más altos estándares de seguridad. Normalmente, las vacunas destinadas a niños se prueban primero en adultos, para después ir poco a poco probándolas en niños y bebés. Los ensayos clínicos se llevan a cabo en varias fases que pueden progresar secuencialmente, pero que también se pueden superponer en determinadas circunstancias. Estas fases son:
– Fase 1. El objetivo durante esta primera fase es obtener la mayor cantidad de información acerca de la seguridad de la vacuna. Generalmente ésta se administra a entre 20 y 100 voluntarios que no han estado expuestos previamente a la enfermedad en cuestión y que están sanos. Estos primeros estudios se utilizan para determinar si hay reacciones adversas con el aumento de las dosis y, de ser posible, para obtener información temprana sobre la eficacia de la vacuna para inducir una respuesta inmunitaria en las personas.

– Fase 2. Si durante la fase 1 no se han observado problemas de seguridad, la vacuna pasa a inocularse a más personas, normalmente entre 100 y 300. Estos cientos de voluntarios se escogen de manera que sus características físicas (sobre todo edad) sean parecidas a las de las personas que van a recibir la vacuna, pero que tengan estados de salud diferentes en cada caso (algunos sanos, otros con algún achaque, otros enfermos, etc..). Se escogen también personas de diversos orígenes y grupos demográficos para garantizar la representación de diferentes poblaciones. En esta segunda fase se prueban diversas dosis en estudios aleatorizados y controlados. Estos estudios proporcionan información adicional sobre la seguridad de los efectos secundarios y riesgos comunes a corto plazo, también sobre la relación entre la dosis administrada y la respuesta inmunitaria, y proporcionan información inicial sobre la eficacia de la vacuna para generar una respuesta inmunitaria en humanos. Se utilizan pruebas estandarizadas y validadas para evaluar la respuesta inmunitaria. Estos estudios de vacunas suelen incluir un grupo de control compuesto por personas que pueden recibir una vacuna ya aprobada por la EMA o la FDA, un placebo u otra sustancia. Las personas que reciben la vacuna en estudio se comparan con las del grupo de control.
– Fase 3. La vacuna se administra generalmente a miles de personas (entre 1000 y 3000) y el estudio genera información crucial sobre su eficacia y datos adicionales de seguridad importantes. Esta fase incluye información adicional sobre la respuesta inmunitaria y compara a quienes reciben la vacuna con el grupo de control. Por ejemplo, se compara el número de casos de la enfermedad en el grupo vacunado con el del grupo de control para determinar si la vacuna reduce la incidencia de la enfermedad. Estos estudios también proporcionan información sobre la seguridad de la vacuna, incluyendo la identificación de efectos secundarios menos comunes.

• El proceso de fabricación de la vacuna. Durante los ensayos clínicos de fase 3, la EMA o la FDA examina el proceso de fabricación de la vacuna propuesto por la institución que la va a producir. También se inspeccionarán las instalaciones de donde se fabricará la vacuna para garantizar que cuenten con todo lo necesario para una producción a gran escala fiable y consistente. Se producen en condiciones reales varios lotes de vacuna que se someten a una serie de pruebas para garantizar la consistencia de la vacuna entre diferentes lotes. Las agencias reguladoras exigen a los fabricantes que presenten los datos de estas pruebas para respaldar un proceso de fabricación exitoso, incluso después de la aprobación.
Si el Comité Asesor de Vacunas y Productos Biológicos Relacionados exige cambios en alguno de los procesos de fabricación o no ve claros algunos datos, se ponen en contacto con los fabricantes para hacer las modificaciones pertinentes.
• Solicitud de aprobación. Una vez que se ha desarrollado un proceso de fabricación que garantice que la vacuna se pueda producir de forma fiable y consistente, y completados con éxito los programas de desarrollo preclínico y clínico, las empresas presentan una Solicitud de “Licencia” a las agencias reguladoras. En EE. UU. esta solicitud se denomina Solicitud de Licencia de (productos) Biológicos (Biologics License Application, BLA) a la FDA. Una BLA es una solicitud exhaustiva que se presenta a la Agencia y que incluye datos e información preclínicos y clínicos, así como detalles del proceso de fabricación y las instalaciones. La FDA examina la BLA en detalle analizando los datos de los ensayos clínicos para determinar si los resultados demuestran que la vacuna es segura y eficaz. La BLA también contiene información de prescripción, que incluye información sobre el uso, la dosis y la administración de la vacuna, todo ello basado en datos científicos. A partir de aquí, la FDA decide si aprueba el uso de la vacuna. La FDA toma sus decisiones basándose en el análisis de los beneficios y riesgos para la población que recibirá la vacuna. El equipo científico de la FDA trabaja en colaboración para evaluar todos los datos e información científica incluidos en la BLA y toma la decisión de aprobar o no una vacuna.
• Actuación del comité asesor. En el caso de EE. UU., en algunos casos, antes de la aprobación de una vacuna, un comité de la federal independiente denominado Comité Asesor de Vacunas y Productos Biológicos Relacionados (Vaccines and Related Biological Products Advisory Committee, VRBPAC) puede aportar información sobre datos científicos para evaluar la seguridad, la eficacia y el uso de la vacuna. El VRBPAC está formado por expertos científicos y de salud pública independientes que debaten la vacuna propuesta en un foro público. Si este comité exige cambios en alguno de los procesos de fabricación o no ve claros algunos datos, se ponen en contacto con los fabricantes para hacer las modificaciones pertinentes.

• Aprobación de la vacuna. Las agencias reguladoras analizan miles de páginas de datos e información de fabricación como parte de una solicitud. Si los datos son convincentes y determinan que la vacuna es segura y eficaz para el uso previsto, que sus beneficios superan los riesgos para las personas que probablemente la recibirán y que el proceso de fabricación garantiza la calidad y la consistencia del producto, la agencia autorizará la vacuna. En el caso de la FDA, su aprobación implica que una empresa puede comercializar en EE.UU. la vacuna para su uso en la población para la que está aprobada.
• Información de prescripción y etiquetado. Con base en los datos científicos presentados en la solicitud, las agencias reguladoras revisan y determinan si la información de prescripción refleja de forma adecuada y precisa las indicaciones, el uso, la dosis y la administración aprobados. La información de prescripción se actualiza según sea necesario para incluir la información más reciente sobre la vacuna, disponible y revisada por las agencias.
• Fabricación a gran escala. Después de que una vacuna recibe la licencia, una empresa puede empezar a producir lotes más grandes para distribuirla al público. Esto no quiere decir que la vigilancia del cumplimiento de los más estrictos estándares de seguridad se abandone, al contrario, las agencias reguladoras continuarán supervisando las actividades de producción de vacunas, lo que incluye inspecciones periódicas de las instalaciones de fabricación para garantizar el cumplimiento de las regulaciones impuestas. Esto continuará mientras el fabricante tenga una licencia para la vacuna.
• Monitorización de la seguridad de las vacunas. Las agencias, en colaboración con otras instituciones académicas y grandes sistemas de atención médica no gubernamentales, utilizan sistemas de vigilancia pasiva y activa para supervisar la seguridad de las vacunas tras su aprobación. En EE. UU. estos sistemas incluyen el Sistema de Notificación de Eventos Adversos de Vacunas (Vaccine Adverse Event Reporting System, VAERS), el programa BEST (Biologics Effectiveness and Safety, Eficacia y Seguridad de Productos Biológicos) y Sentinel de la FDA, la colaboración de la FDA con los Centros de Servicios (médicos) de Medicare y Medicaid (CMS) para evaluar las reclamaciones médicas y los datos proporcionados por los Centros para el Control y la Prevención de Enfermedades (Centers for Disease Control and Prevention’s, CDC).
• Fase 4 de ensayos clínicos. En algunos casos las agencias reguladoras exigen que un fabricante realice estudios posteriores a la comercialización, los denominados ensayos clínicos de Fase 4 para evaluar con más detalle los riesgos graves conocidos o potenciales de la vacuna tras ser administrado a miles de personas. La fase 4 es un estudio detallado y continuo para evaluar la seguridad y la eficacia de la nueva vacuna a lo largo de un período más largo.
El proceso de fabricación de las vacunas es muy largo y lleno de controles estrictos en cada fase para evitar cualquier error. A su vez, el control posterior del uso de una vacuna nos asegura que no pueda haber ningún tipo de riesgo para la población.
• Fabricación de nuevos lotes. Cada lote de vacunas no puede ser distribuido hasta que las agencias reguladoras lo analicen y lo autoricen. Para autorizar un lote de vacuna, la agencia revisa los resultados de las pruebas del fabricante, que generalmente incluyen la esterilidad, pureza, eficacia y consistencia de la vacuna, y puede realizar pruebas de confirmación.
• Seguimiento continuo del uso de las vacunas. Las agencias reguladoras controlan continuamente el uso de las vacunas. Este control es fundamental para que las agencias puedan proporcionar una regulación eficaz de las vacunas. Los científicos de las agencias realizan diversas investigaciones que contribuyen a la formulación de políticas, evaluaciones de riesgos, nuevos métodos y estándares, y cambios en el etiquetado de productos, incluyendo la promoción de nuevas técnicas para evaluar la seguridad, potencia y eficacia de las vacunas, además de estrategias para el desarrollo de nuevas vacunas.
Como podemos ver, el proceso de fabricación de las vacunas es muy largo y lleno de controles estrictos en cada fase para evitar cualquier error. A su vez, el control posterior del uso de una vacuna nos asegura que no pueda haber ningún tipo de riesgo para la población.
Pero, sabiendo ahora cómo es el proceso de preparación y que éste puede durar muchos años, ¿cómo es posible que la vacuna del COVID19 se aprobara en tan poco tiempo? ¿Se saltaron pasos que la hacen menos segura? ¿Fueron los controles de seguridad más laxos? Para responder a estas preguntas debemos dejar atrás las teorías conspirativas y las respuestas son muy claras: la vacuna del COVID19 siguió las mismas fases de fabricación que una vacuna normal y es tan segura como cualquier otra vacuna. Veamos como sucedió todo.
En diciembre de 2019, en concreto el 31 de este mes, China notifica los primeros casos de neumonía por un virus desconocido en Wuhan. El 7 de enero de 2020 se identifica el virus y se averigua que es un nuevo coronavirus que se denomina SARS-CoV-2. Un coronavirus es un tipo de virus que pertenece a una gran familia llamada Coronaviridae que se caracterizan por su forma esférica y la presencia de proyecciones en su superficie que se parecen a una corona solar, de ahí su nombre (“corona” en latín). Son muy comunes y hay una gran variedad de ellos. Pueden causar infecciones respiratorias y digestivas en humanos y animales. En humanos, van desde enfermedades leves hasta graves. Son causantes de los casi inofensivos pero molestos resfriados comunes, pero también causan enfermedades más graves como el Síndrome Respiratorio Agudo Severo (SARS, causados por el virus SARS-CoV), Síndrome Respiratorio de Oriente Medio (MERS, causado por el virus MERS-CoV) o la famosa COVID-19 (causada por el virus SARS-CoV-2).
Un avance importante se produjo el 11 de enero de 2020, cuando China publica el genoma completo del virus. Esto permite iniciar el diseño de vacunas a nivel mundial. Una cosa común y corriente, que pasa inmediatamente después de que se conoce un nuevo virus, es que las grandes compañías farmacéuticas comienzan a estudiarlo. Su intención es sacar una vacuna cuanto antes pues esto les reportará beneficios económicos importantes si resulta que el virus se convierte en un problema. En este caso, la situación no fue diferente y el mismo enero empresas como Pfizer/BioNTech o Moderna comenzaron a realizar ensayos preclínicos con vacunas que preparaban con la tecnología del ARN mensajero (ARNm). Estos primeros estudios que se realizan en animales ya muestran una respuesta inmunitaria prometedora. La cuestión en este caso fue que ya desde muy al principio expertos de muchos países avisaron que este virus iba a causar un problema grave de salud pública y cuando esta ya fue una realidad y medio mundo ya estaba confinado en su casa, comenzaron los ensayos clínicos. Para el 16 de marzo de 2020 la empresa americana Moderna inicia el primer ensayo clínico en humanos, la fase 1, con su vacuna mRNA-1273. A esta le siguen BioNTech y Pfizer que comienzan estudios similares en abril con su vacuna BNT162b1 (luego BNT162b2).

Vista la gravedad de la pandemia mundial y que los resultados de la Fase 1 estaban siendo satisfactorios, comienza la fase 2 en mayo de 2020. Esta fase ya cuenta con cientos de voluntarios y se observa que la vacuna genera anticuerpos y es bien tolerada. Para julio del 2020 se inicia la fase 3 con decenas de miles de voluntarios en múltiples países: el estudio de Pfizer/BioNTech cuenta 43000 participantes, el de Moderna con 30000 y el de AstraZeneca con más de 30000. Se realiza un seguimiento a estos voluntarios durante meses, pero en octubre/noviembre ya se han recopilado suficientes datos como para analizar la eficacia de la vacuna. El 8 de noviembre de 2020 Pfizer/BioNTech anuncia que su vacuna tiene más del 90% de eficacia en prevenir la COVID-19 sintomática y el 16 de noviembre: Moderna reporta una eficacia del 94.5%. Una vez publicados estos datos y en manos de la EMA y la FDA, se trabaja a contrarreloj en estas agencias para evaluar si deben ser aprobadas o no. La aprobación llega al mes siguiente, en diciembre de 2020 cuando se autoriza el uso de emergencia de varias vacunas: Reino Unido aprueba la vacuna de Pfizer/BioNTech (BNT162b2) el 2 de diciembre, el 11 de diciembre la FDA aprueba la vacuna de Pfizer, el 18 diciembre hace lo propio con la de Moderna y el 30 de diciembre la EMA aprueba la de Pfizer/BioNTech. De inmediato, estas vacunas comienzan a ser inoculadas a la gente. A partir de enero de 2021 da comienzo la fase 4 en la que se monitorea de manera continua de seguridad y eficacia a gran escala en la población general.
Como se puede ver, la vacuna del COVID19 tardó menos de 12 meses en ser aprobada desde su fase preclínica. Esto hace que haya comentarios en contra de la seguridad de las vacunas contra la COVID-19 argumentando que “se hicieron demasiado rápido” y “seguro se saltaron pasos”. Pero en realidad, no se saltó ninguna de las fases clínicas obligatorias. Lo que sí ocurrió fue una aceleración sin precedentes del proceso gracias a factores logísticos, científicos y financieros. Pero ¿cómo se consiguió y por qué con esta vacuna en particular?
La vacuna del COVID19 siguió las mismas fases de fabricación que una vacuna normal y es tan segura como cualquier otra vacuna.
Bien, para entender esto lo primero que debemos tener en cuenta es que estábamos inmersos en una pandemia mundial con millones de personas confinadas en sus casas, miles muriendo a diario y la economía mundial en una recesión casi sin precedentes. Estas circunstancias extraordinarias llevaron a tomar medidas también extraordinarias. ¿Cómo se pudo ir más rápido, pero sin saltarse pasos? Veámoslo.
Para empezar, las diferentes fases de los ensayos clínicos se llevaron a cabo en paralelo. Normalmente, las fases I, II y III se hacen de forma secuencial, esperando meses o años entre una y otra, pero en el caso del COVID-19 se solaparon fases. Por ejemplo, fase II empezó antes de completar la fase I, basándose en datos preliminares seguros; y Pfizer, Moderna y AstraZeneca realizaron ensayos de Fase I/II combinados, aprobados por agencias reguladoras. Esto, aunque no muy común, no es algo sin precedentes, ya que ya había ocurrido en otras ocasiones con otras vacunas.

Por otro lado, y, de nuevo, dadas las extraordinarias circunstancias del momento, se produjo una inversión económica masiva. ¿Por qué? Porque las economías se estaban hundiendo y había que salir de la situación como fuera. Por ello, era más “barato” financiar con miles de millones una vacuna que esperar a que la situación se solucionara con el tiempo y perder así mucho más de lo invertido. Gobiernos como el de EE. UU., Reino Unido y la Unión Europea financiaron con miles de millones de euros el desarrollo y la producción anticipada, incluso asumiendo el riesgo de que las vacunas fracasaran y se perdiera la inversión. Esto permitió a las farmacéuticas fabricar millones de dosis antes de que la vacuna estuviera aprobada, de nuevo, a riesgo de perder esas dosis. En condiciones normales, las empresas no asumen ese riesgo.
Otra cuestión importante que explica el rápido desarrollo de las vacunas contra el COVID19 fue que la tecnología necesaria ya estaba desarrollada. Pfizer y Moderna llevaban más de 10 años investigando las vacunas de ARN mensajero (ARNm) para desarrollar vacunas contra otros virus como el Zika, el MERS o la rabia. Esto significa que no se partió desde cero, sino que se adaptó la tecnología existente al SARS-CoV-2.
Se puede afirmar que aunque el proceso fue excepcionalmente rápido, NO se omitió ninguna fase crítica. Si se pudo acelerar el proceso fue gracias a avances tecnológicos previos, financiación inmediata y en cantidades elevadísimas, gran cantidad de voluntarios y una coordinación de entre agencias, empresas y gobiernos sin precedentes.
También jugó un papel fundamental el hecho de que la gente de a pie estaba también deseosa de salir de la situación dada las grandes restricciones de las libertades individuales que había en aquel momento. Esto ocasionó que hubiera una altísima cantidad de voluntarios para probar las vacunas. Más de 70.000 personas participaron en los ensayos de Fase III de Pfizer, Moderna y AstraZeneca. Este alto número de personas inoculadas, más el hecho de que el virus estaba circulando de forma rapidísima y descontrolada, hizo que fuera posible detectar eficacia en pocos meses por la cantidad de casos. En otras vacunas, con enfermedades menos prevalentes, se necesitan años para tener resultados estadísticamente sólidos.
Por último, debemos tener en cuenta que en las circunstancias de aquel momento no había otras cosas más importantes y la implicación de las agencias reguladoras en el proceso fue intensa y continua. Las agencias realizaban un control casi diario de los resultados y, en esta ocasión, en lugar de esperar a que la farmacéutica entregara todos los datos al final, la FDA, EMA y otras agencias revisaban los datos de manera continua (“rolling review” en inglés). De este modo pudieron autorizar de manera rápida pero informada, en cuanto los datos clave (eficacia, seguridad, calidad) estuvieron disponibles. Esto no quiere decir que los estándares regulatorios se modificaran, para nada. Si que es cierto que la aprobación se basó en los estándares de emergencia, pero los requisitos para autorizar una vacuna en uso de emergencia fueron claros y públicos desde el principio: eficacia ≥50%, datos de seguridad de al menos 2 meses posteriores a la última dosis y evaluación de riesgos frente a beneficios. Estos estándares se aplicaron estrictamente y no se rebajaron.
En definitiva, se puede afirmar vehementemente que, aunque el proceso fue excepcionalmente rápido, NO se omitió ninguna fase crítica. Si se pudo acelerar el proceso fue gracias a avances tecnológicos previos, financiación inmediata y en cantidades elevadísimas, gran cantidad de voluntarios y una coordinación de entre agencias, empresas y gobiernos sin precedentes. De hecho, se puede concluir, que estas vacunas siguen siendo las más monitoreadas en la historia en términos de eventos adversos, lo que fortalece su perfil de seguridad.
